
Chemical Engineering
Our chemical property prediction software predicts all relevant thermodynamic properties for any chemical compound with high accuracy. This allows chemical engineers to work with compounds even when no measured data is available.
Prediction Accuracy
The following sections compare predicted vapor liquid equilibirum data for pure compounds and mixtures with the corresponding experimental reference data. Note that our software allows the prediction of many more properties not mentioned here. For details on all available features take a look at the shop.
Pure Properties
The prediction of pure properties is especially important when no measured data is available for the compound of interest. Knowledge of the vapor liquid equilibrium (VLE) of a pure compound is essential for many separation problems. Below we show a comparision of predicted VLE data T(pre) to thermodynamic reference data T(ref):
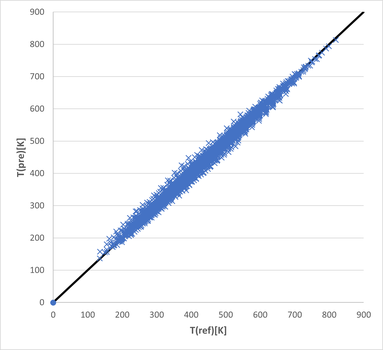
Deviation in Temperatures:
The mean value of the absolute deviation in temperature is 6.4 K. The coefficient of determination is 0.992. These are incredibly small deviations for the broad range of compounds and temperatures included.
Deviation in Volumes:
The mean value of the absolute relative deviation in volumes is 3.5%. Again the deviation from the reference data is very small. Such accurate volume predictions show a high potential for the prediction of transport properties like viscosities in future releases.
The reference data is generated by the PC-SAFT equation using parameters fitted to experimental data. This way a broad range of temperature and pressure conditions are included. More than 4500 datapoints for different compounds are shown. The deviations of the reference data from the original measured data is negligible.
A broad range of functional groups are represented by the data above including: Alkanes, Alkenes, Alkynes, Cyclics, Multicyclics, Aromatics, Halogenes, Aldehydes, Ketones, Ethers, Esters, Maleates, Alcohols, Organic Acids, Nitriles, Nitrates, Amines, Thioethers, Silanes, Halosilanes, Organosilianes and Multifunctional-Compounds. For all these compounds the same set of parameter is used to translate the data of quantum mechanic predictions into equation of state parameter. There are no group specific corrections necessary. Therefore this model has a much more universial character than typical group contribution methods and is applicable to any molecule. For an overview of the methodology applied please read the following article: J. Phys. Chem. B 2008, 112, 5693-5701.
A further advantage of the presented model is given by the used equation of state. The PC-SAFT equation is know for its ability to accurately inter- and extrapolate thermodynamic data even if only few refererence datapoints are available. The data shown below uses a single experimental datapoint to improve the prediction accuracy. For consistency this datapoint is always the VLE data at 0.7 * Tc. Tc is defined as the critical temperature of the compound.
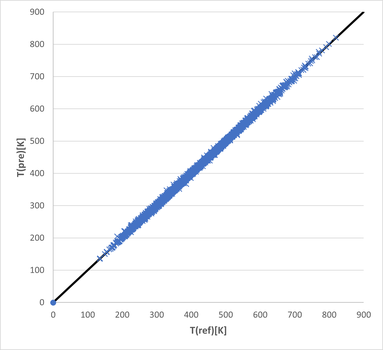
Deviation in Temperatures:
The mean value of the absolute deviation in temperature is reduced to 1.6 K over the whole temperature range of all components. The coefficient of determination is 0.999. Such small deviations are unachieved by any other predictive approach know to us.
Deviation in Volumes:
The mean value of the absolute relative deviation in volumes is 0.3% and thereby often negligible for practical applications.
It is advised to use available pure component experimental data whenever possible to drastically increase the prediction accuracy. For most compounds at least one measured pure component datapoint is available (i.e. normal boiling point or critical temperature and pressure). An accuracy boost as shown above will therefore be possible in most cases.
Mixture Properties
A property of major interest for mixtures are binary vapor liquid equilibria (VLE). The knowlege of this property is essential in most separation processes. Two examples of fully predicted binary VLE's are shown below. The experimental data is reproduced with minor deviations.

VLE of n-decane / ethane. Comparision of the predicted values to experimental data.
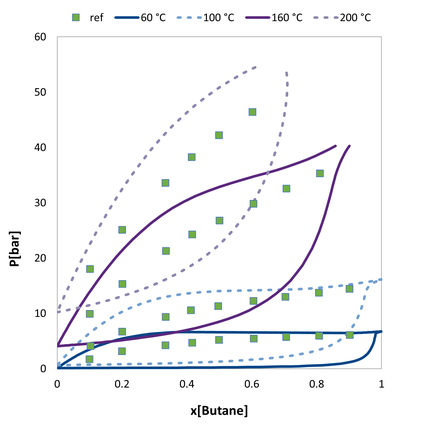
VLE of 1-butanol / n-butane. Comparision of the predicted values to experimental data.
Some predictions may show larger deviations than shown above. The main reason for larger deviations are inaccuracies of the pure component vapor pressure prediction. A single pure component datapoint is often already sufficient to increase the accuracy significantly, as shown below. For consistency the pure component VLE data at 0.7 * Tc is used again.
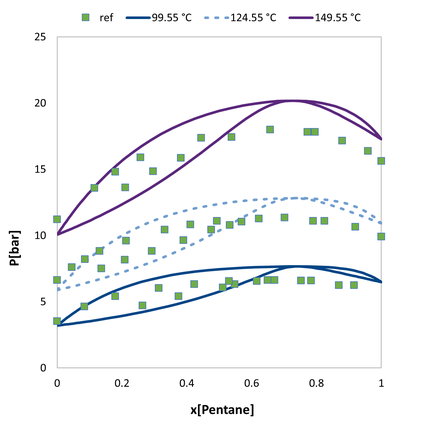
VLE of the acetone / n-pentane. Comparision of the predicted values to experimental data.
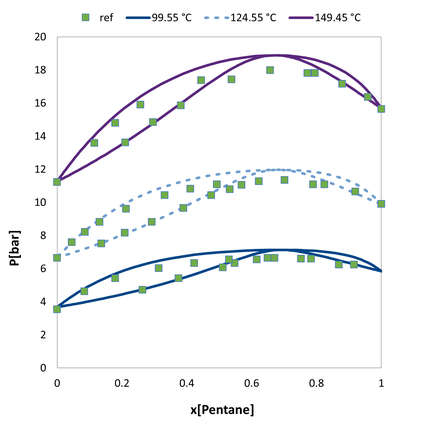
VLE of the acetone / n-pentane. Comparision of the predicted values augmented with a single pure compound datapoint to experimental data.

VLE of the n-butane / carbon dioxide. Comparision of the predicted values to experimental data.
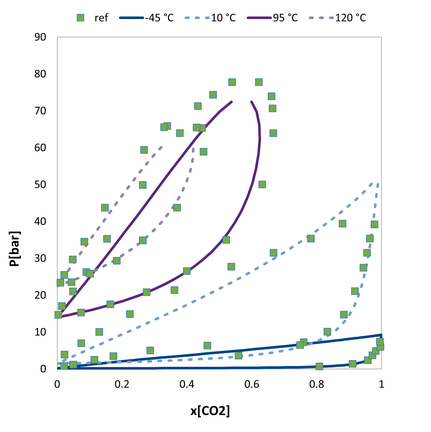
VLE of the n-butane / carbon dioxide. Comparision of the predicted values augmented with a single pure compound datapoint to experimental data.
The results clearly indicate a significant improvement in accuracy. The prediction accuracy further increases with the amount pure component data added to the simulation. Note that the pure component data not only improve the starting and end point of the binary VLE, but also increase the prediction accuracy of the mixture properties within the VLE. The adjusted pure component parameter directly affect the mixing contributions within the equations.
We currently use the GFN-xTB methodology for the quantum mechanic predictions. This results in very short simulation times with a good prediction accuracy. In future releases other quantum mechanical methods will also be supported. This results in longer simulation times, but allows even higher accuracy in predictions.